
成果简介
相关工作以《Single-atom Mo-tailored high-entropy-alloy ultrathin nanosheets with intrinsic tensile strain enhance electrocatalysis》为题在《Nature Communications》上发表论文。
图文导读
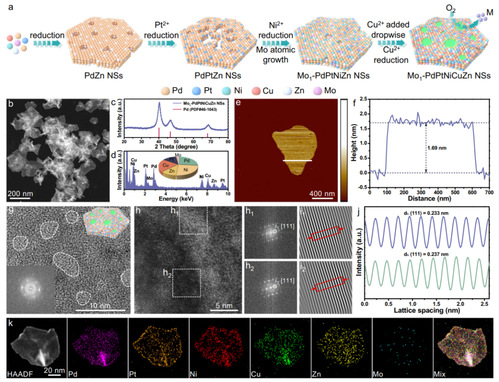
图1 Mo1-PdPtNiCuZn SAHEA NSs的合成与表征
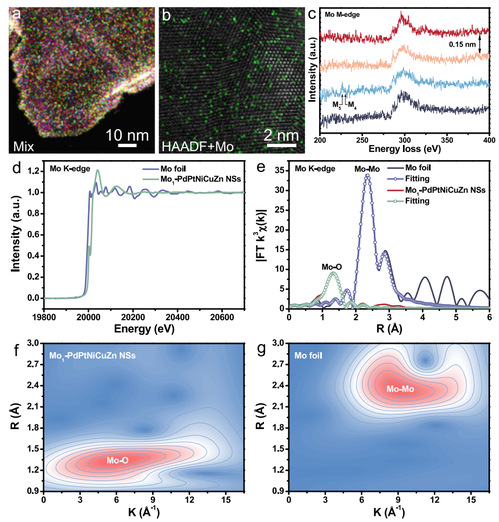
图2 Mo1-PdPtNiCuZn SAHEA NSs的组成及电子结构表征
XANES光谱显示,Mo1-PdPtNiCuZn SAHEA NSs的Mo的K边吸附阈值位置和白线强度均高于Mo箔,表明表面暴露的Mo原子处于高价态,与XPS结果一致(图2d)。值得注意的是,Mo1-PdPtNiCuZn SAHEA NSs的XANES光谱在前边缘区域显示20010 eV附近有一个肩峰,表明存在具有畸变[MoO6]八面体结构的MoOx。对应的FT-EXAFS谱图显示,Mo1-PdPtNiCuZn SAHEA NSs的主峰位于~1.33 Å,这归因于MoOx在SAHEAs中的Mo-O散射。Mo-Mo散射的特征峰几乎不存在,这有力地证实了Mo1-PdPtNiCuZn SAHEA NSs中的Mo主要以原子分散位点的形式存在,进一步表明Mo是表面修饰物质。
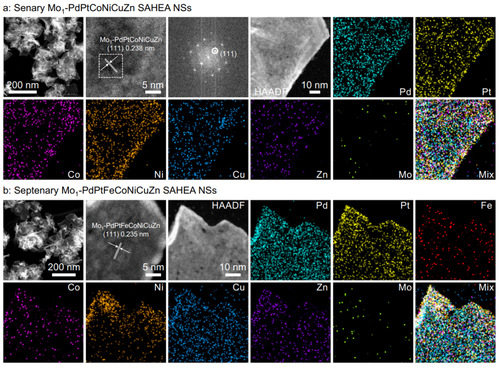
图3 SAHEA NSs合成的通用性
同时,在上述所有的SAHEA NSs中都观察到明显的本征拉伸应变、晶格变形和缺陷,这主要是由于超薄的特性和各原子尺寸的显著差异造成的。这些结果证实了本方法合成单原子Mo定制SAHEA NSs的通用性。此外,发现Cu(acac)2的加入是形成Mo1-PdPtNiCuZn SAHEA NSs的关键。在其他条件不变的情况下,在反应前同时引入Cu(acac)2和其他金属前驱体,可以得到Mo1-PdPtNiCuZn SAHEA纳米粒子(NPs)。

图4 不同催化剂在碱性电解质中的MOR性能
在N2饱和的0.1 M HClO4溶液中,观察到CV曲线具有典型的氢吸附/解吸峰和Pt氧化/还原峰,表明这些催化剂具有较高的Pt原子利用率(图4a)。值得注意的是,在Mo1-PdPtNiCuZn SAHEA NSs和Mo1-PdPtNiCuZn SAHEA NPs的放大循环伏安曲线中,可以明显地识别出Mo的氧化峰(~1.21 V),进一步证明了Mo原子的存在。
为了评估MOR稳定性,对Mo1-PdPtNiCuZn SAHEA NSs、Mo1-PdPtNiCuZn SAHEA NPs、PdPtNiCuZn HEA NPs和商用Pt/C催化剂在0.77 V下进行了10000 s的计时电流测量。如图4所示,Mo1-PdPtNiCuZn SAHEA NSs、Mo1-PdPtNiCuZn SAHEA NPs和PdPtNiCuZn HEA NPs都显示出比商用Pt/C高得多的电流密度,表明它们具有优异的MOR长期耐久性。值得注意的是,Mo1-PdPtNiCuZn SAHEA NSs的催化活性可以在连续五个循环的计时电流测试后通过更换新的电解质来恢复,并显示出良好的再现性(图4e)。
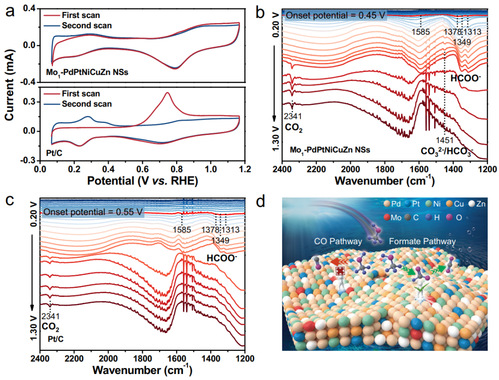
图5 MOR机理
为了深入了解Mo1-PdPtNiCuZn SAHEA NSs高MOR性能的来源,采用CO溶出和原位光谱测试进行分析。如图5a所示,与Pt/C相比,Mo1-PdPtNiCuZn SAHEA NSs在0.75 V附近的氧化峰受到更大的抑制,表明其对CO的耐受性显著增强。这是因为Mo1-PdPtNiCuZn SAHEA NSs中的多种金属可以显著稀释Pt-Pt位点,从而在表面形成孤立的Pt位点,并且拉伸应变扩大了Pt-Pt距离。这些结构特征有利于MOR路径切换为无CO主导途径,因为形成负载需要至少三个连续的Pt原子或缺陷位点的存在。此外,亲氧Mo单原子的暴露和修饰可以有效地调节其相邻位点的d带中心,从而在反应物的有效解离和中间体的适当结合之间实现良好的平衡,从而提高MOR选择性和加速反应动力学。本研究采用原位FTIR对MOR电催化过程中的反应中间体进行了跟踪。在所研究的电位窗口中,可以清楚地观察到2400-1200 cm-1之间的几个特征峰(图5b、c)。具体而言,约1585、1378和1349 cm-1的峰分别归因于甲酸盐(HCOO-)的vas(OCO)、δ(C-H)和vs(OCO)。位于1313 cm-1左右的峰属于甲酸盐的另一个特征峰。值得注意的是,在整个电位范围内没有COL信号,而CO2(约2341 cm-1)出现在高电位,表明CO2来源于弱吸附甲酸盐的进一步氧化。
结合CO剥离和原位FTIR研究发现,Mo1-PdPtNiCuZn SAHEA NSs可以避免COads的形成,并将MOR转换为甲酸为主的途径(图5d),而Pt/C的MOR过程同时涉及CO途径和甲酸途径,导致催化剂失活。
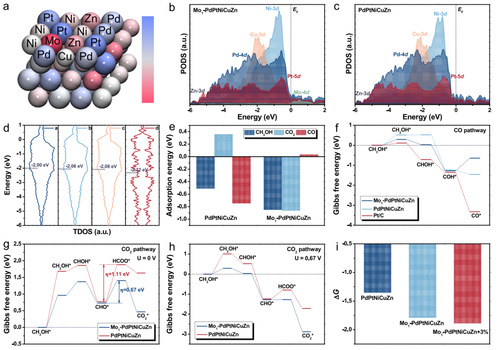
图6 DFT计算
通过DFT计算,进一步验证了Mo1-PdPtNiCuZn SAHEA NSs的MOR性能提高的机理。考虑Mo单原子的掺入和拉伸应变的引入,构建了Mo1-PdPtNiCuZn、Mo1-PdPtNiCuZn和PdPtNiCuZn的应变表面作为计算模型。Bader电荷模拟表明,随着电子密度的增大,Pt原子的电荷态变得更负,表明电子更多分布在Pt原子周围,而Mo原子是缺电子的(图6a)。结果表明,引入单原子Mo可以有效地优化Pt位点周围的电子密度。
为了进一步了解Mo1-PdPtNiCuZn SAHEA和PdPtNiCuZn HEA的电子结构,绘制了HEA中各元素的PDOSs图。如图6b、c所示,Ni和Mo元素在MOR过程中充当电子耗尽中心,促进HEA表面的电子转移。同时,Pt、Pd、Cu和Zn之间有效的d-d轨道耦合不仅降低了氧化过程中电子转移的能垒,而且稳定了Pt位点的价态,有利于MOR中间体的稳定。与PdPtNiCuZn相比,在Pt位点附近引入Mo单原子进一步提高了电活性。此外,计算了不同模型的总d态密度图,以显示d带中心,其位置可以直接与增强的MOR活性相关联(图6d)。因此,d带中心上移的Mo1-PdPtNiCuZn与关键反应中间体和*OH的结合能更强,从而增加了这些中间体进一步氧化成CO2的机会。
本文进一步从能量角度比较了Mo1-PdPtNiCuZn和PdPtNiCuZn的性能。如图6e所示,单原子Mo修饰的PdPtNiCuZn对CH3OH和CO2的吸附更强,导致对MOR的电活性增强。正如预期的那样,Mo1-PdPtNiCuZn表现出非优先吸附CO。可以推断,在碱性电解质中,Mo1-PdPtNiCuZn SAHEA NSs几乎不吸附CO分子。这些计算表明,HEA的形成显著稀释了连续的Pt-Pt位点,而Mo单原子作为促进剂的植入有效地调节了HEA宿主的电子结构,从动力学和热力学上都阻止了COads的形成,从而避免了CO中毒,将反应同步切换到甲酸为主的途径。
原文链接:https://www.nature.com/articles/s41467-024-45874-z
(来源:粉末冶金及硬质合金展)